《单基因糖尿病知多少?》
接《如何看待“4岁女孩爱吃甜食确诊糖尿病”这新闻?》
单基因糖尿病(Monogenic diabetes)是由单个基因或染色体位点的一个或多个缺陷引起。该疾病可能在家族内作为显性、隐性或非孟德尔性状遗传,但也可能由于新发突变而自发出现。
单基因糖尿病是一大类少见疾病的组合,约占儿童糖尿病的2.5%–6.5%。然而,笔者注意到很多单基因糖尿病被误诊、漏诊,所以今天就来说说它。
本篇文章内容主要参考:国际儿童青少年糖尿病学会(The International Society for Pediatric and Adolescent Diabetes, ISPAD)的《诊断和治疗单基因糖尿病的2022年共识》
(即《ISPAD Clinical Practice Consensus Guidelines 2022: The diagnosis and management of monogenic diabetes in children and adolescents》)。
一,什么情况下提示单基因糖尿病?
在儿童中,大多数单基因糖尿病病例是由导致β细胞丢失或功能障碍的基因突变引起的,很少部分是由导致非常严重的胰岛素抵抗的基因突变引起。
从临床角度来看,应考虑单基因糖尿病诊断的具体情况包括:
①糖尿病在 6 个月大之前出现,这被称为 新生儿糖尿病(NDM)。
②常染色体显性遗传、家族性轻度高血糖或糖尿病。
③伴有如下特征的儿童糖尿病:先天性心脏或胃肠道缺陷、脑畸形、严重腹泻或非常年幼儿童的其他自身免疫性疾病。
④单基因 胰岛素综合征的提示信号;这些信号来自于高胰岛素水平或高胰岛素需求。比如,脂肪分布异常,皮下脂肪缺乏,尤其是四肢; 血脂异常,尤其是高甘油三酯;和/或严重的黑棘皮症。
二,在出生的6个月-12个月诊断出糖尿病
在出生6个月-12个月时被诊断糖尿病的大多数患者是1型糖尿病。但如下信息提示单基因糖尿病:
①1型糖尿病相关的自身抗体阴性;
②不寻常的家族史
③有如下临床问题:大舌±发育迟缓±脐带缺陷±先天性心脏病、胰腺发育不全(脂肪泻)、肝功能异常、面部畸形、自身免疫性肠炎、骨骼发育不良、肠道畸形、多种自身免疫性疾病(免疫性溶血、甲状腺疾病等)
三,KATP通道基因 (KATP-NDM) 突变引起的永久性新生儿糖尿病
KATP-NDM 是永久性新生儿糖尿病(PNDM) 的最常见原因,也是短暂性新生儿糖尿病(TNDM) 的第二大最常见原因。
KATP-NDM 在特定群体中的患病率取决于血缘关系的程度。在近亲结婚的后裔中,PNDM 最常见的已知原因是 KATP 通道或 INS 基因异常。 如果父母有亲属关系,Wolcott-Rallison 综合征或 GCK 基因纯合突变是最常见的病因。 另外,高达 20% 的 PNDM 病例的原因仍然未知。
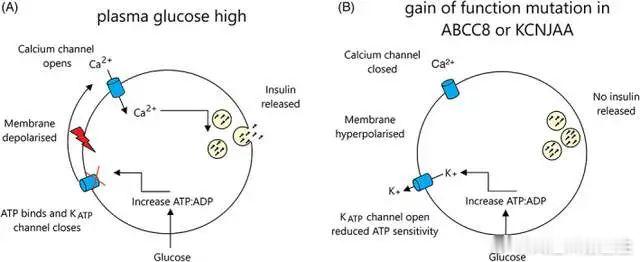
KATP 通道是由四个形成孔的 Kir6.2 亚基和四个 SUR1 调节亚基形成的异源八聚体复合物,分别由基因 KCNJ11 和 ABCC8 编码。 它们通过将细胞内代谢状态与β细胞膜电活动联系起来来调节胰岛素分泌。细胞内代谢活动的任何增加都会诱导胰腺β细胞内 ATP/ADP 比率的增加。高 ATP/ADP 比率关闭 KATP 通道并导致细胞膜去极化,最终触发胰岛素分泌。
激活 KCNJ11 或 ABCC8 突变,阻止 KATP 通道关闭,从而减少响应高血糖的胰岛素分泌,导致糖尿病。
图1:胰腺 β 细胞在 (A) 高血糖环境中的正常细胞和 (B) 在 K-ATP 通道突变的细胞中分泌胰岛素。
大约 90% 的 KCNJ11 突变患者患有 PNDM,而 ~10% 的患者发展为 TNDM,而 ABCC8 突变更频繁 (~66%) 导致 TNDM。 两种 NDM 亚型之间 IUGR 的严重程度或糖尿病诊断年龄没有显著差异。
KATP 通道突变通常显示较轻的子宫内发育迟缓,并且诊断时间略晚于 6q24 异常的婴儿,表明在宫内发育的最后几个月和出生时胰岛素缺乏不太严重。在 KATP-TNDM 中,糖尿病通常比 6q24-TNDM 缓解更晚,复发更早。
KATP-NDM 中 C 肽水平低或检测不到,并且经常出现糖尿病酮症酸中毒,表明胰岛素依赖。
除糖尿病外,约 20% 的 KCNJ11 突变患儿表现出相关的神经系统问题。这是因为神经系统细胞也需要KATP 通道维持正常功能。
破坏性最严重的突变也与明显的发育迟缓和早发性癫痫有关,称为 DEND(发育迟缓、癫痫和 NDM)综合征。以 NDM 和较轻的发育迟缓为特征的中等 DEND 综合征更常见,但无癫痫。
最近利用详细测试的研究表明,即使在那些以前被认为只会导致孤立性糖尿病的较轻突变中,也会发生轻度神经发育异常。比如,这些儿童中的许多符合发育性协调障碍(特别是视觉空间运动障碍)、注意力缺陷多动障碍、焦虑障碍或自闭症的标准,和/或有行为或睡眠困难。
磺脲类(SU)药物与胰岛β细胞膜上的 磺脲类受体(SUR1亚基) 结合,导致 KATP通道关闭。 KATP通道关闭后,细胞内钾离子外流减少,细胞膜去极化。 去极化激活 电压依赖性钙通道(VDCC),钙离子内流增加。 钙离子浓度升高触发 胰岛素囊泡释放,从而促进胰岛素分泌。
SU不仅仅可以治疗KATP-NDM的高血糖,还可以治疗神经症状。比如,ABCC8 编码的 SUR-1 蛋白在视网膜功能中至关重要,SU (格列本脲) 通过 SUR-1 介导的机制赋予直接视网膜神经保护。 但因为血脑屏障的存在,要更好疗效或需要其他药物。
四,胰岛素原基因 (INS) 突变引起的新生儿糖尿病
INS基因突变是继 KATP 通道突变之后 PNDM 的第二大常见原因。
显性杂合突变最常见,通常导致胰岛素原分子发生错误折叠,该分子被困并积聚在亚细胞区室中,导致内质网应激和 ß 细胞凋亡。
隐性双等位基因 (纯合子或复合杂合子) 突变导致胰岛素原丢失或失活。这些突变不会导致缓慢进行性β细胞破坏,但会导致出生前后胰岛素生物合成不足,这解释了受影响儿童出生体重低得多和糖尿病表现较早的原因。
杂合子 INS 突变儿童的子宫内发育迟缓的严重程度与 KATP 通道突变儿童相似,但它们出现的年龄稍晚。
虽然这类儿童的糖尿病最常在 6 个月大之前被诊断出来,但它也可能发生在一岁甚至更晚。因此,对于早期出现的自身抗体阴性糖尿病儿童,应考虑进行基因检测。再就是一些具有 MODY 样表型的儿童也需要考虑。
大多数杂合子 INS 突变是散发的新生突变,但约 20% 的先证者有常染色体显性遗传 NDM 家族史。
五,Wolcott-Rallison 综合征
Wolcott-Rallison 综合征 (WRS) 是由 EIF2AK3(真核翻译起始因子 α 2-激酶 3)基因的双等位基因突变引起的。其特征是早发性糖尿病、脊椎骨骺发育不良、复发性肝和/或肾功能障碍。
该基因编码一种参与调节内质网 (ER) 应激反应的蛋白质。在没有功能性蛋白的情况下,胰腺发育是相当正常的,但错误折叠的蛋白质在出生后积累在内质网内,最终诱导β细胞凋亡。
虽然糖尿病通常在婴儿期表现出来,但可能要到 3-4 岁才会出现。糖尿病可能是该综合征的首发临床表现,因此,即使在孤立性 PNDM 的儿童中也应考虑这种诊断,特别是如果他们是近亲生育人群。
由于该病是隐性遗传的,因此兄弟姐妹的复现该病的风险为 25%。暴发性肝功能衰竭是 WRS 患者死亡的主要原因,目前没有药物可以逆转这种异常。肝脏(有或没有胰腺)移植可以挽救生命并改善患有这种综合征的人的预后。
六,GCK 突变引起的新生儿糖尿病
葡萄糖激酶被认为是 β 细胞的葡萄糖传感器,因为它催化葡萄糖磷酸化的限速步骤,因此使 β 细胞能够对血糖程度做出适当的反应。
继发于两个等位基因突变(纯合子或复合杂合子)的完全葡萄糖激酶缺陷会阻止β细胞分泌胰岛素以响应高血糖。
新生儿表现为严重的子宫内发育迟滞,通常在出生后的最初几天被诊断为糖尿病,需要外源性胰岛素治疗。除糖尿病外,他们没有表现出任何相关的胰腺外特征。
GCK 占 PNDM 总病例的 2%-3%,但在血缘关系高度的地区患病率增加。 这种类型的 PNDM 以隐性方式遗传,因此未来兄弟姐妹的复现该病的风险为 25%。
该类PNDM孩子的父母往往是无症状的轻度血糖增高。因此,通常建议测量任何 NDM 患儿的父母的空腹血糖,即使没有已知的糖尿病。
患有 KATP/PNDM 或胰岛素基因 (INS) 异常的个体似乎也不容易出现严重的糖尿病眼部并发症。
(未完待续)