导语
转录组多组学在免疫系统中的应用研究旨在通过分析和解读基因的转录过程,揭示免疫系统中的基本机制、调控网络和功能特征。这种研究方法结合了转录组学、蛋白质组学和其他多组学技术,能够提供全面的关于免疫应答和免疫系统疾病的信息。
免疫系统是机体对抗感染和疾病的重要组成部分,涉及到许多免疫细胞、信号通路和分子机制。通过转录组学的高通量测序技术,可以识别和分类各类免疫细胞,以及不同亚群之间的基因表达差异,进一步了解免疫系统的多样性和分化过程。
通过分析差异表达基因和共表达网络,可以识别免疫调控关键基因、信号通路和转录因子,深入了解免疫应答的调控机制。比较免疫疾病患者和健康个体的基因表达,可以鉴定出与免疫相关的基因和生物标志物,为免疫疾病的诊断、治疗和预后提供新的靶点和指标。揭示药物作用的分子机制,并指导个体化治疗策略的设计。
总之,转录组多组学在免疫系统中的应用研究为我们提供了全面的视角,帮助我们理解免疫系统的复杂性和调控机制。这种综合性的研究方法为免疫相关疾病的预防、诊断和治疗提供了新的思路和策略。
文献一:激活 NRF2 可重塑氧化代谢缺陷,从而恢复慢性阻塞性肺病的巨噬细胞功能

文献标题:NRF2 Activation Reprograms Defects in Oxidative Metabolism to Restore Macrophage Function in Chronic Obstructive Pulmonary Disease
发表期刊:American Journal of Respiratory and Critical Care Medicine
影响因子:24.7
发表时间:2023.4
文章链接:DOI: 10.1164/rccm.202203-0482OC
研究背景
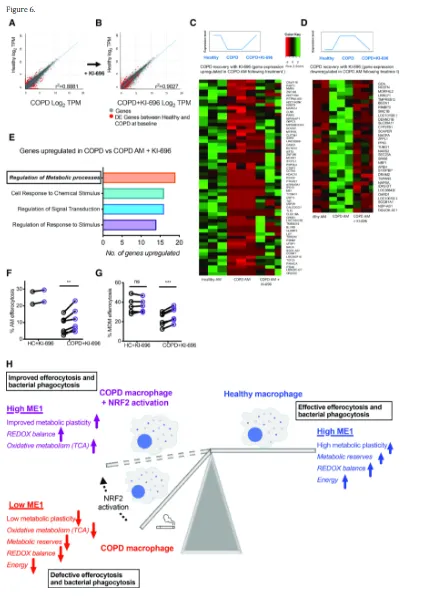
慢性阻塞性肺疾病(COPD)是一种以持续性气道炎症和巨噬细胞功能紊乱为特征的疾病。巨噬细胞生物能量学的改变在多大程度上导致抗氧化反应受损和疾病发病机制尚未完全确定。目标:通过对慢性阻塞性肺病肺泡巨噬细胞(AMs)和外周单核细胞衍生巨噬细胞(MDMs)的研究,我们试图确定核心代谢过程中的内在缺陷是否会导致巨噬细胞功能障碍和氧化还原失衡。方法:来自慢性阻塞性肺病供体和健康供体的AM和MDM进行了功能、代谢和转录分析。测量和主要结果:我们观察到,来自COPD供体的AM和MDMs显示出糖酵解和线粒体呼吸衍生的能量储备的临界消耗,并且过度依赖糖酵解作为ATP的来源,导致能量状态降低。氧化代谢缺陷延伸到与NADPH生成酶ME1(苹果酸酶1)表达缺陷相关的氧化还原平衡受损,ME1是抗氧化转录因子NRF2(核因子红细胞2相关因子2)的已知靶标。因此,NRF2的选择性激活重置了COPD转录组,导致TCA循环中间体的产生增加、能量状态改善、良好的氧化还原平衡和巨噬细胞功能的恢复。结论:在慢性阻塞性肺病中,代谢可塑性的固有丧失会导致代谢耗竭和氧化还原能力降低,这可以通过激活NRF2通路来挽救。因此,通过NRF2增强靶向这些缺陷,可能为治疗COPD中描述的异常气道炎症提供一种有吸引力的治疗策略。
文献二:针对新辅助化疗诱导的胰腺癌代谢重编程可促进抗肿瘤免疫和化疗反应

文献标题:Targeting neoadjuvant chemotherapy-induced metabolic reprogramming in pancreatic cancer promotes anti-tumor immunity and chemo-response
发表期刊:Cell Reports Medicine
影响因子:14.3
发表时间:2023.10
文章链接:DOI: 10.1164/rccm.202203-0482OC
研究背景
胰腺导管腺癌(PDAC)在化疗下的分子动力学仍不完全清楚。新辅助化疗(NAC)的广泛使用为研究化疗后的PDAC样本提供了独特的机会。利用来自复旦大学上海癌症中心的队列,包括暴露于和未暴露于新辅助白蛋白结合型紫杉醇和吉西他滨(AG)的PDAC样本,我们汇编了来自单细胞和空间转录组、蛋白质组、批量转录组和代谢组的数据,加深了我们对化疗后PDAC分子变化的理解。代谢通量分析显示,NAC诱导PDAC代谢模式的重编程并增强免疫原性。值得注意的是,NAC导致糖酵解的下调和CD36的上调。组织微阵列分析表明,CD36高表达与接受术后AG的患者生存率较差有关。靶向CD36可协同改善体外和体内对AG的PDAC反应,包括患者来源的临床前模型。

文献三:经典猪瘟病毒感染过程中自噬对细胞稳态和抗病毒先天免疫的调控

文献标题:The regulation of cell homeostasis and antiviral innate immunity by autophagy duringical swine fever virus infection
发表期刊:Emerging Microbes & Infections
影响因子:13.2
发表时间:2023.12
文章链接: DOI: 10.1080/22221751.2022.2164217
研究背景
CSFV(经典猪瘟病毒)目前在亚洲发展中国家流行,最近在日本重新出现。在自然选择压力的压力下,CSFV不断进化,以维持其在自然界中的生态位。CSFV已经进化出诱导免疫抑制的机制,但其致病机制尚不清楚。在这项研究中,使用转录组学和代谢组学方法,我们发现CSFV感染通过激活干扰素通路、抑制宿主炎症、细胞凋亡和重塑猪肺泡巨噬细胞中的宿主代谢来改变先天宿主免疫。此外,我们发现自噬可以改变CSFV感染诱导的先天免疫和代谢。增强的自噬进一步抑制了CSFV诱导的RIG-I-IRF3信号转导轴和JAK-STAT信号通路,阻断了I型干扰素的产生,同时减少了CSFV感染细胞中NF-κB信号通路和细胞凋亡的自噬抑制。此外,CSFV感染诱导的糖酵解水平和乳酸和丙酮酸的含量以及3-磷酸甘油醛(糖酵解转化为丝氨酸的衍生物)通过自噬改变。我们还发现,沉默糖酵解代谢的限速酶HK2(己糖激酶2)可以诱导自噬,但会降低干扰素信号通路、NF-κB信号通路,并抑制CSFV感染诱导的细胞凋亡。此外,通过沉默ATG5或使用3-甲基腺嘌呤抑制细胞自噬,可以回填沉默HK2对细胞干扰素信号通路、NF-κB信号通路和细胞凋亡的抑制作用。

参考文献
Ryan EM, Sadiku P, Coelho P, Watts ER, Zhang A, Howden AJM, Sanchez-Garcia MA, Bewley M, Cole J, McHugh BJ, Vermaelen W, Ghesquiere B, Carmeliet P, Rodriguez Blanco G, Von Kriegsheim A, Sanchez Y, Rumsey W, Callahan JF, Cooper G, Parkinson N, Baillie K, Cantrell DA, McCafferty J, Choudhury G, Singh D, Dockrell DH, Whyte MKB, Walmsley SR. NRF2 Activation Reprograms Defects in Oxidative Metabolism to Restore Macrophage Function in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2023 Apr 15;207(8):998-1011. doi: 10.1164/rccm.202203-0482OC. PMID: 36724365; PMCID: PMC7614437.
Tang R, Xu J, Wang W, Meng Q, Shao C, Zhang Y, Lei Y, Zhang Z, Liu Y, Du Q, Sun X, Wu D, Liang C, Hua J, Zhang B, Yu X, Shi S; Chinese Study Group for Pancreatic Cancer. Targeting neoadjuvant chemotherapy-induced metabolic reprogramming in pancreatic cancer promotes anti-tumor immunity and chemo-response. Cell Rep Med. 2023 Oct 17;4(10):101234. doi: 10.1016/j.xcrm.2023.101234. PMID: 37852179; PMCID: PMC10591062.
Li X, Song Y, Wang X, Fu C, Zhao F, Zou L, Wu K, Chen W, Li Z, Fan J, Li Y, Li B, Zeng S, Liu X, Zhao M, Yi L, Chen J, Fan S. The regulation of cell homeostasis and antiviral innate immunity by autophagy duringical swine fever virus infection. Emerg Microbes Infect. 2023 Dec;12(1):2164217. doi: 10.1080/22221751.2022.2164217. PMID: 36583373; PMCID: PMC9848339.
