文:回溯档案
编辑:回溯档案
马铃薯疮痂病 (CS) 是一种由致病性链霉菌引起的难治性疾病,在世界范围内发生,但人们对马铃薯疮痂病与土壤微生物组之间的相互作用知之甚少。

在本研究中,对自然高和低结痂严重程度,我们的目的是确定与马铃薯 CS 相关的土壤微生物组的组成和假定功能。
链霉菌菌株的分离与鉴定
进行纯培养实验,利用琼脂培养基平板上的富集培养法和稀释法从马铃薯病斑中分离出链霉菌菌株,将病灶匀浆,与9 mL无菌水混合,室温孵育30 min,用无菌水稀释10倍和100倍,然后涂在燕麦琼脂平板,28°C 培养 3-7 天。选择单菌落形成单位,在OMA平板上传代3次,用细菌通用引物27F和1492R扩增分离菌株的16S rRNA基因。

PCR 扩增子在 1% 琼脂糖凝胶上进行验证,反应产物在 ABI 3730XL DNA 分析仪上进行纯化和测序,通过核苷酸 BLAST 以确定菌株的大致系统发育关系,如果 NCBI BLAST 报告的序列的最大同一性 > 97% 并且是所有列出的匹配中最大的,则分类学得到确认,通过MEGA7软件使用邻接法构建系统发育树以可视化系统发育关系。
理化分析
在三个技术重复中测定了 ZS 和 FS 的理化特性,以评估研究领域的土壤条件,使用土壤和不含CO 2的去离子水以1:2.5的比例的混合物测量土壤pH,土壤总碳、有机质、全氮、铵、硝酸盐、速效磷、速效钾、有效硫是根据之前的研究确定的。

根据制造商的说明,使用 ENZA? 土壤 DNA 试剂盒提取 GS、RS、ZS 和 FS DNA,并使用 NanoDrop 2000 分光光度计测定 DNA 的数量和质量,DNA 储存于 ? 80 °C 直至进一步分析。
定量PCR
在三个技术重复中通过 qPCR 检测细菌 16S rRNA 基因和 thaxtomin 生物合成基因txtAB拷贝数,引物 515F 和 806R 用于扩增来自细菌的 16S rRNA 基因的 292 bp 片段 ,引物 StrepF和 StrepR用于扩增来自疮痂病链霉菌的 thaxtomin 生物合成基因txtAB的 72 bp 片段。
使用 96 孔板在 CFX96? 实时系统上进行分析,扩增最终体积为 20 μL,含有 10 μL SYBR? Premix Ex Taq?、5 μL DNA、0.2 μL 各 10 μM 引物和 4.6 μL ultra-纯净水。构建含有16S rRNA基因片段或txtAB基因片段的质粒以制备各自的标准曲线,并使用在线计算器自动计算质粒拷贝数基于碱基对的浓度、大小和平均重量。
通过实时荧光 qPCR 后对已知拷贝数的上述质粒进行系列稀释,生成显示基因拷贝数与 Ct 值之间关系的标准曲线方程,在相同的 PCR 运行后,根据标准曲线方程和样品自身的 Ct 值计算样品的拷贝数,每次 PCR 均包括无模板对照以及已知 Ct 值的阳性对照。

细菌16S和txtAB基因扩增的热循环条件为95°C 5分钟;95℃10s、55℃10s、72℃20s循环40个;以及最终的熔化循环,以产生用于质量控制的熔化曲线,所有运行都有r的标准效率曲线2 ?> 0.99,效率为 90–110%。
Illumina MiSeq 测序和分析
引物 341F和 805R用于扩增 16S rRNA 基因的 V3-V4 高变区,根据制造商的建议,使用 NEB Next? Ultra? DNA Library Prep Kit for Illumina构建扩增子文库,并添加索引代码,扩增子文库在 MiSeq PE250 测序仪上进行测序,并生成 250 bp 的双端读数。

Illumina 测序后生成大约 2.2 GB 原始读数,然后使用 USEARCH v. 9.2 软件按操作分类单元对所得配对序列读数进行合并、修剪、过滤、比对和聚类 ,合并配对序列读数后,总共生成 1,878,784 个合并序列,每个土壤样本平均有 48,174 个序列。
USEARCH中的UPARSE-OTU算法将相似度≥97%的序列分配给同一个OTU,并在OTU聚类时使用cluster_otus命令过滤嵌合体,质体和线粒体的序列以及未分类在细菌域中的序列被丢弃,所有样品中序列号为 1 的 OTU 也被丢弃。

使用 USEARCH 推荐的数据库进行分类分配:16S 的 RDP 训练集,α 和 β 多样性指数是根据每个样本 29,718 个序列深度的稀有 OTU 表计算的,Alpha 多样性指数,包括 ACE、Chao1、覆盖率、Shannon 和 Simpson 指数,上述数据处理的命令行可在附加文件 2中找到。
鸟枪法宏基因组测序和分析
基于上述16S扩增子测序结果,选择GS DNA进行鸟枪法宏基因组测序,以更高分辨率评估微生物群落组成和功能,根据制造商的说明,使用 TruSeq? DNA PCR-free 样品制备试剂盒构建宏基因组文库。

宏基因组文库在 HiSeq 2500 测序仪上进行测序,并生成 150 bp 的双端读数,Illumina 测序后生成约 244.4 GB 原始读数,所有 10 个 GS 样本总共产生 727,482,738 个配对序列读数。
使用一系列软件程序对所得序列读数进行质量控制分析:SeqPrep用于在读数中触发非生物碱基,例如引物或条形码,以及 Sickle用于过滤tripping后长度小于50 bp且平均质量得分小于20的reads。

每个样本获得52.4-8050万个干净的reads,基于de Bruijn图进行de novo组装以获得contigs,总共生成了1,088,639个contigs,选择长度 > 500 bp 的重叠群来使用 MetaGene 预测开放阅读框 ,获得1,731,093个ORF,所有预测的基因都使用 CD-HIT 进行成对比对。
其中 90% 以上的长度可以与另一个具有 95% 以上同一性的基因比对,作为除最长基因之外的冗余被删除,从而形成由 1,205,798 个基因组成的非冗余基因目录,平均长度为 579.02 bp。

将基因目录翻译的推定氨基酸序列与 NCBI-NR 数据库中的蛋白质/结构域进行比对,使用 NR 数据库中相应的分类信息对基因进行分类注释。在每个样本中,每个分类单元的映射读数被计为分类单元映射读数的数量。
从原始鸟枪法宏基因组测序读数中分析了微生物群落的组成,随后将细菌分类学分析与微生物分类学分析分开,微生物分类学分析的相对丰度通过 MetaPhlAn v. 2.0 参考数据库的独特进化枝特异性标记基因进行量化,然后按比例标准化为值,使用Zhang描述的方法计算细菌类群的估计绝对丰度。

低痂严重程度组的特征是 GS 微生物组具有低txtAB基因丰度、低细菌丰度和高多样性
通过测定 ZS 和 FS 的理化特性来评估研究田的土壤条件,本研究的理化分析表明,CS发生在酸性土壤条件下,ZS和FS的pH值范围为4.32至6.81,进行双尾 Wilcoxon 秩和检验以确定 H 和 L 中物理化学特征之间的差异。
ZS中铵(NH 4 + -N)的浓度显着不同,H 组和 L 组之间,但 FS 中所有观察到的理化特征没有显着差异,除铵态氮外,土壤TC和OM的浓度?也存在显着差异,组合 ZS 和 FS 时的 H 和 L 组。

TxtAB是赤霉病植物毒素 ThxA 生物合成的关键基因,能够准确定量致病性链霉菌菌株,为了确定赤霉病病原体丰度,使用所有土壤样品检测thaxtomin生物合成基因txtAB的拷贝数,但只有GS在实时PCR系统中产生清晰的荧光信号。
表明GS含有较高的大量的结痂病原体,而其他样品含有很少或不含结痂病原体,此外,痂病病原体的基因txtAB拷贝数显着GSH 和 GSL 之间存在差异,并且与结痂严重程度显着正相关。

在高和低结痂严重程度之间,GS 中观察到细菌群落组成存在显着差异
双尾Wilcoxon秩和检验用于评估组间细菌相对丰度在属水平上的差异,与PCoA结果一致,?不同土壤区划之间存在最大数量的分化属,RSL和FSL之间总共发现了143个分化细菌属,GSH和FSH之间总共发现了132个差异细菌属。
GSL和RSL之间发现了53个分化细菌属,而GSH和RSH之间只发现了7个分化细菌属,这表明H可能减少GS和RS之间的群落差异,在四个土壤-根系系统区室中,GS 包含 58 个分化的H 和 L 之间的细菌属,远大于 RS、ZS 和 FS 中分别鉴定的 1、5 和 7,这一发现进一步表明 GS 的细菌群落组成与 CS 的严重程度相关。

低结痂严重程度组的特点是 GS 微生物群具有低致病性链霉菌丰度
微生物群落内的相对丰度通过 MetaPhlAn v. 2.0 软件进行量化,与 NR 比对结果类似,微生物群落由细菌、真核生物和病毒组成,作为微生物群落的主要组成部分,细菌被分离出来,宏基因组细菌群落中的两个链霉菌属S . 酸疥疮和S . turgidiscabies是可能的痂病病原体。
与 NR 对齐结果类似,S.酸疥疮是我们样品中 MetaPhlAn2 分析的主要赤霉病病原体,在我们的培养实验中,11 个分离菌株与S具有最高的系统发育相似性,Acidicabies菌株中,1 与S具有最高的相似性。

turgidiscabies菌株根据NCBI BLAST结果;使用MEGA7软件构建的系统发育树来可视化系统发育关系,培养实验进一步证实了S的存在,酸疥疮和S .turgidiscabies并鉴定出S。酸疥疮是本研究中主要致病性链霉菌属。

GS微生物组的群落组成和功能与CS相关
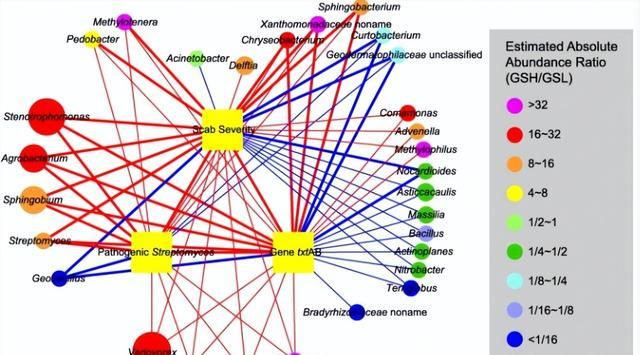
根据宏基因组数据,细菌群落由116个属组成,为了明确GS中CS敏感属,选择三个参数,包括痂病严重程度、致病性链霉菌的EAA和txtAB基因拷贝数,并使用Spearman与细菌属EAA的相关性分析进行分析。
三个参数彼此之间呈显着正相关,28 个属显着相关与结痂严重程度分级,其中 17 例呈阳性,11 例呈阴性,16个属与致病链霉菌?的EAA呈显着相关,其中13个属呈阳性,3个属呈阴性,27个属与txtAB基因拷贝数显着相关,其中16个属阳性,11个属阴性,与多个参数相关的微生物被认为是最受关注的,共有 13 个属与?所有 3 个参数均显着正相关。
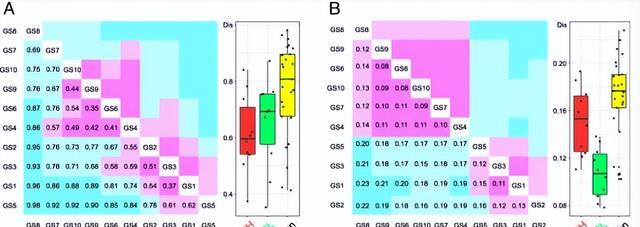
计算基于Bray-Curtis排序的相异性度量来量化宏基因组细菌群落组成和KO功能类别的组成相异性,我们的宏基因组功能分析共产生了3328个KO功能类别,并且在微生物群落组成和功能方面,GSH和GSL之间的差异大于组内差异 ;群落组成,P ?= 6.3 × 10 ?13 ;群落功能,表明 GSH 和 GSL 之间的群落组成和功能均不同。
群落功能的相异系数值低于群落组成的相异系数,表明GS的微生物功能比其群落组成更相似,特别是,GSL的群落功能表现出显着的差异性低于GSH,而GSL的细菌群落组成则表现出略高差异,换句话说GSL 的微生物功能谱比 GSH 更相似,尽管其群落组成的差异性稍高,总体而言,结痂严重程度与 GS 内微生物功能的关系比与群落组成的关系更密切,并且 L 样本的功能相似性高于 H 样本。
使用双尾 Wilcoxon 检验对 GSH 和 GSL 之间的功能差异进行量化和可视化,在?GSH 和 GSL 中分别发现了240 个和 561 个显着富集的 KO 功能类别,为了阐明哪些 KO 功能类别是这些差异中最主要的,相对丰度大于 0.03% 的差异 KO 功能 类别b. 通过该过滤器,保留了 98 个差异 KO 功能类别,其中 16 个在 GSH 中显着富集,其中 82 个在 GSL 中显着富集。
K02035的相对丰度在所有差异KO功能类别中最高,K00799次之;它们都富含谷胱甘肽,K02035是一种肽/镍转运系统底物结合蛋白,是“QS”途径的成员,K00799被认为参与“谷胱甘肽代谢”和“异生素”生物降解和代谢”途径。

在GSL、K03046中富集的差异KO功能类别中, DNA 指导的 RNA 聚合酶亚基 β是最丰富的 KO 功能类别,其次是 K03043,这些功能类别主要涉及“嘌呤代谢”、“嘧啶代谢”和“RNA聚合酶”途径,此外,我们还计算了涉及丰富差异KO功能类别的途径,GSH和GSL均以“代谢”途径为主要组成部分,但GSL中有多种属于“遗传信息加工”的途径,而GSH则没有此类途径。

结语
本研究旨在解决土壤微生物组与CS之间的相互作用,并证实GS的微生物群落组成和功能与CS严重程度相关。低文本ABGS 中的基因丰度、低细菌丰度、高多样性、高共现网络复杂性和高群落功能相似性是马铃薯 CS 严重程度低的指标。
与高赤霉病严重程度的马铃薯相关的 GS 微生物组包含致病性相关基因谱,以及涉及“ABC 转运蛋白”、“细菌分泌系统”、“QS”、“氮代谢”途径以及细胞色素 P450 的一些代谢的多个基因H组中显着富集,相比之下,一些抗生素的生物合成途径在L组中显着富集。
【1】Kim YC、Leveau J、Gardener BBMS、Pierson EA、Pierson LS、Ryu CM。植物相关细菌促进植物健康的多因素基础。应用环境微生物。2011;77(5):1548–55。
【2】Berg G. 植物-微生物相互作用促进植物生长和健康:农业中控制使用微生物的前景。应用微生物生物技术。2009;84(1):11-8。
【3】刘K,纽曼M,麦金罗伊JA,胡CH,克勒珀JW。用于多种植物病害生物防治的植物促生长根际细菌的选择和评估。植物病理学。2017;107(8):928–36。
【4】Santhanam R、Weinhold A、Goldberg J、Oh Y、Baldwin IT。与根部相关的原生细菌可以拯救植物免受连作期间出现的突然枯萎病的侵害。国家科学院院刊。2015;112(36):E5013–20。
【5】徐晓明,Jeffries P,Pautasso M,Jeger MJ。结合使用生物防治剂来管理植物病害的理论和实践。植物病理学。2011;101(9):1024–31。
