研究背景
神经节细胞胶质瘤是一种分化良好、生长缓慢的胶质神经元肿瘤,由发育不良的神经节细胞和肿瘤胶质细胞组成。好发于儿童和青年,多位于颞叶,与癫痫发作有关。也可发生于任何年龄段和神经轴内的任何部位,包括小脑、脑干和脊髓。影像学表现多样,但通常表现为实性成分和囊性成分的混合。大多数神经节细胞胶质瘤在组织学符合WHO 1级,完全切除后不会复发。部分肿瘤呈间变性特征,术后出现复发。
BRAF癌基因的p.V600E热点突变已在神经节细胞胶质瘤中被发现,突变的发生率约为10%-60%,其中大脑皮质肿瘤的发生频率最高,脊髓肿瘤的发生频率较低。然而,BRAF p.V600E突变并不是神经节细胞胶质瘤所特有的,可见于多种神经上皮肿瘤,包括毛细胞性星形细胞瘤、胚胎发育不良性神经上皮瘤(DNET)、儿童IDH野生型弥漫性星形细胞瘤、青少年多形性低级别神经上皮瘤(PLNTY)、多形性黄色星形细胞瘤和上皮样胶质母细胞瘤。此外,BRAF p.V600野生型神经节细胞胶质瘤的基因改变尚不明确,本研究对40例经病理学证实的神经节细胞胶质瘤进行了全面的分子分析,以评估该肿瘤的遗传学特征。
研究结果
(1)人口学和临床特征
本研究纳入了40名经病理证实的神经节细胞胶质瘤患者(表1、表S2)。其中男23例,女17例,年龄0~64岁(中位年龄21岁)。肿瘤位于大脑半球31例(78%),其中颞叶19例,额叶3例,顶叶4例,枕叶5例。肿瘤位于小脑4例,丘脑2例,脊髓3例。首次手术治疗后的临床随访期为0~29年(中位数为1.8年)。26例患者达到肿瘤全切,其中2例在1.2年和7.8年后复发。11例患者达到次全切除,其中4例肿瘤复发(0.6至10年后)。3名患者的切除程度未知,其中2名患者在1.4年和1.8年后肿瘤进展。
(2)组织病理特征
所有肿瘤都混合有畸形的神经节细胞和肿瘤性胶质细胞成分。胶质细胞成分呈星形细胞形态37例(93%),少突胶质细胞形态3例(8%)。27例(68%)可见嗜酸性颗粒体,6例(15%)可见Rosenthal纤维。19例(48%)有钙化,其中8例存在广泛钙化。19例肿瘤中有16例存在CD34阳性分支细胞(84%)。所有肿瘤均无间变性特征,如高有丝分裂指数(每10个高倍视野中2个以上)、坏死或微血管增生。
(3)遗传学改变

图1:40例神经节细胞胶质瘤患者的肿瘤遗传学汇总表,包括患者年龄、性别、肿瘤位置、基因改变、肿瘤细胞数目、染色体改变
研究者对40个神经节细胞胶质瘤进行了二代测序,对基因突变、基因融合、扩增、缺失和染色体拷贝数改变进行评估。27例肿瘤中存在BRAF癌基因的致病性改变,其中18个为p.V600E热点突变,5个为非V600E突变(其中2个p.R506delinsRVLR,p.L505delinsLEYLS、p.R506delinsRSTQ、p.T599_W604delinsTDG各1例),以及4个基因框内融合(in-frame gene fusions)(2个以KIAA1549为融合伴侣,1个与KLHL7融合,1个与CDC42BPB融合)。在13个无BRAF改变的肿瘤中,9个存在预测可以激活MAP激酶信号通路的其他基因改变:其中2个存在KRAS p.Q61K热点突变,1个存在ERC2-RAF1基因框内融合,1个存在FGFR1热点错义突变(p.N546K),1个存在FGFR1-TACC1基因框内融合,1个存在影响FGFR2基因第17外显子剪接受体序列的突变,2个存在FGFR2基因框内融合(分别以INA和KIAA1598为融合伴侣)。1例临床诊断为1型神经纤维瘤病的患者存在中存在NF1基因种系杂合性移码突变。这些涉及BRAF、KRAS、RAF1、NF1、FGFR1和FGFR2的基因改变是相互排斥的(即没有肿瘤同时存在任何两种变异)。总体而言,在40个肿瘤中,有36个(90%)确认含有导致MAP激酶信号通路激活的基因改变。在其余4个肿瘤中,3个无可识别的致病性改变,1个存在新发现的ABL2-GAB2基因融合,已在儿童白血病中发现。
3例存在BRAF p.V600E突变的神经节细胞胶质瘤同时存在CDKN2A纯合性缺失,其中1例肿瘤还同时存在PTEN亚克隆错义突变。除此之外,在所有肿瘤中没有发现额外的致病性突变、融合、扩增或缺失。因此,在33个肿瘤中,BRAF、KRAS、RAF1、NF1、FGFR1或FGFR2改变是孤立的致病性改变。
染色体拷贝数分析显示,在26个肿瘤中,没有染色体的获得、丢失或局部扩增或缺失。在另外14个肿瘤中,每个肿瘤的染色体改变数量从1至10。在大多数情况下,染色体拷贝数的变化仅限于整个染色体或染色体臂的获得或缺失,除了3个肿瘤存在CDKN2A基因纯合性缺失外,没有发现局部扩增或纯合性缺失。频繁出现的染色体拷贝数改变包括7号染色体三体型(含BRAF基因座)6个,5号染色体三体型5个,12号染色体三体型3个,9号染色体单体3个,1p单体3个。在6个带有7三体的肿瘤中,有4个是含有BRAF改变的肿瘤,可能涉及突变或融合。3例9号染色体单体肿瘤均累及9p21,导致CDKN2A基因纯合性缺失。3例1p单体型肿瘤均含有BRAF p.V600E突变。
(4)遗传学改变与临床和影像学之间的关系
遗传学改变与确诊年龄无明显相关性。在解剖部位方面,所有存在FGFR改变的神经节细胞胶质瘤都位于大脑半球,而存在BRAF改变的肿瘤位于整个神经轴。4例小脑肿瘤中有3例含有BRAF p.V600E突变,3例脊髓肿瘤中有2例存在KIAA1549-BRAF融合。影像特征包括肿瘤大小、是否存在囊性成分、边界和强化与遗传学改变没有明显的相关性。
(5)遗传学改变与组织病理学的关系
本研究中,3个胶质细胞成分呈少突胶质细胞形态的肿瘤均存在FGFR改变。另外2例存在FGFR改变的肿瘤胶质细胞成分呈星形细胞形态。其他存在BRAF、KRAS、NF1和RAF1改变的肿瘤的胶质细胞成分均为星形细胞形态。其他组织学特征,如嗜酸性颗粒体、Rosenthal纤维、钙化和血管周围淋巴细胞与遗传学改变无明显相关性。
(6)遗传学改变与肿瘤复发或进展的关系
对2例手术全切后复发的肿瘤进行测序分析,其中1例仅存在BRAF p.R506突变,无染色体拷贝数改变。另一例存在BRAF p.V600E突变,CDKN2A纯合性缺失,PTEN抑癌基因错义突变,染色体3q末端获得和9号染色体缺失。在大部切除后进展的4例肿瘤中,3例仅存在BRAF p.V600E突变,无染色体拷贝数改变。第4例存在CDC42BPB-BRAF基因融合。在两例初次手术切除范围不明,后续出现进展的肿瘤中,1例存在FGFR2-INA融合,另一例存在BRAF p.V600E突变。对BRAF改变型与BRAF野生型、BRAF V600E突变与其他BRAF改变、BRAF改变与FGFR改变、BRAF V600E突变/CDKN2A完整与BRAF V600E突变/CDKN2A缺失的患者的无事件生存率进行分析,结果无显著差异。
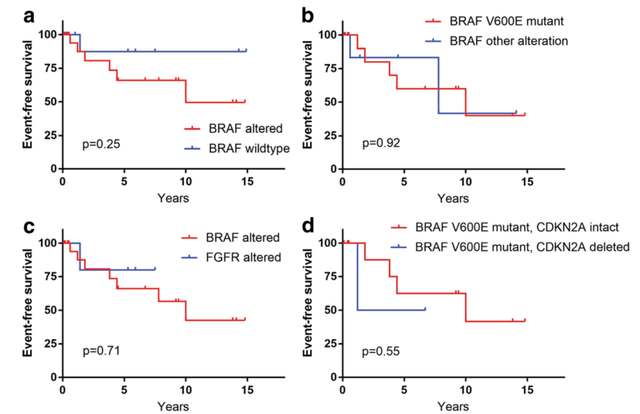
图3:存在不同遗传学改变的肿瘤的无进展生存对比(a:BRAF改变型和BRAF野生型,b:BRAF V600E突变和其他BRAF改变型;c:BRAF改变和FGFR改变型,d:BRAF V600E突变,CDKN2A完整型和BRAF V600E突变,CDKN2A缺陷型)
讨 论
本研究显示,大多数神经节细胞胶质瘤在遗传学层面存在激活MAPK信号通路的改变,主要为BRAF p.V600E突变,以及其他BRAF突变、BRAF融合、FGFR突变或融合、RAF1融合、KRAS突变、NF1突变。上述改变往往是肿瘤存在的唯一基因改变,染色体拷贝数改变较少。这说明神经节细胞胶质瘤是遗传特征较为简单的肿瘤。
神经节细胞胶质瘤与毛细胞星形细胞瘤相比,存在BRAF p.V600E突变或其他BRAF突变的比例更高,后者最常见的是KIAA1549-BRAF融合。神经节细胞胶质瘤存在FGFR1改变的比例较DNET和RGNT低。PLNTY常见BRAF V600E突变或FGFR融合,这与神经节细胞胶质瘤的遗传学改变是重叠的。MVNT常见MAP2K1基因2号外显子的突变或小的框内缺失,以及除了V600E之外的BRAF突变,但神经节细胞胶质瘤不存在MAP2K1基因改变。多形性黄色星形细胞瘤的遗传学特征为BRAF p.V600E突变和CDKN2A纯合性缺失。在本研究中一小部分神经节细胞胶质瘤也存在上述改变。DLGNT的遗传学特征为染色体1p单体型和KIAA1549-BRAF融合。本研究中也发现了3例存在上述改变的神经节细胞胶质瘤,但它们位于成人大脑半球,未见广泛的软脑膜播散。
神经节细胞胶质瘤的遗传特征可以与几种胶质神经元肿瘤区分。本研究中,神经节细胞胶质瘤不存在PRKCA融合或突变,可与乳头状胶质神经元肿瘤和脊索瘤样胶质瘤区分。神经节细胞胶质瘤不存在IDH1/IDH2、TP53、ATRX、TERT启动子、CIC或FUBP1突变,可与成人弥漫性低级别胶质瘤区分。神经节细胞胶质瘤不存在MYB或MYBL1重排,可与血管中心性胶质瘤和儿童IDH野生型弥漫性星形细胞瘤区分。神经节细胞胶质瘤不存在TSC1或TSC2突变,可与室管膜下巨细胞星形细胞瘤区分。皮质发育畸形,包括局灶性皮质发育不良,是神经节细胞胶质瘤的主要鉴别诊断之一。散发局灶性皮质发育不良主要存在PI3K-Akt-mTOR信号通路的改变,如TSC1、TSC2、AKT3、MTOR、PIK3CA或PTEN基因。本研究中仅有1个肿瘤在复发时存在PTEN错义突变,可能是肿瘤进展过程中获得的,而不是驱动肿瘤发生的遗传因素。因此,遗传学检查可能是二者的鉴别要点。
本研究中,4个肿瘤的BRAF基因激酶结构域的β3-αC环第505或506密码子存在框内插入。在COSMIC数据库中的52519个存在BRAF突变的肿瘤中,仅有1个髓母细胞瘤存在该突变。这可能是神经节细胞胶质瘤的新的热点突变。另外,有4个肿瘤未发现MAPK通路相关的改变,可能由于现有技术无法检测这些改变,或存在未知的新改变。其中1个肿瘤存在ABL2-GAB2基因融合,其作用机制尚不明确。
本研究未能发现遗传学特征与疾病进展或复发相关的任何关系。无论是BRAF p.V600E突变或其他改变,最终的结果均是激活Ras-Raf-MEK-ERK通路,所以不同的遗传学改变可能与临床特点或预后无关,而肿瘤位置、切除范围、伴随的其他基因改变或表观遗传学差异更有可能决定患者的临床表现和预后。
参考文献:Pekmezci et al. The genetic landscape of ganglioglioma. Acta Neuropathologica Communications (2018) 6:47
敬请注意:本文仅供相关专业人员学习参考之用,文中的所有信息均不作为诊断和治疗疾病的依据。如出现文中描述的症状,请及时就医。另外,本文仅节选原文的一部分,内容可能不完整或与原文存在偏差,若需更完整的信息请参阅原文。
撰稿:赵赤
审校:张俊平
排版:郜志孟
