导语
16S扩增子测序想必大家都不陌生了,但是拿着一大堆数据和图表,手足无措,不知如何下手,写文章更是没有思路。好多客户来咨询时,常问一个问题是“微生物多样性分析怎么看结果呀”。为解决这一问题,结合元莘生物16S扩增子测序分析报告,我们将开启微生物多样性报告解读专题模块,连载更新16S扩增子测序分析报告,帮助你更好地理解数据,从而更有信心地撰写出高质量的文章。干货满满,特别是没有接触过微生物16s测序的同学非常友好哦~
微生物多样性分析报告主要分为五个部分:OTU聚类和物种注释、Alpha多样性分析、物种组成分析、Beta多样性分析以及差异分析。本期给大家分享的是微生物多样性-OTU聚类和物种注释。
板块一:首先我们需要搞清楚一个非常重要的概念——OTU。
操作分类单位(OTU:Operational Taxonomic Units)是一种操作定义,用于对密切相关的个体群体进行分类。这个术语最初是在1963年由Robert R. Sokal和Peter H. A. Sneath在数字分类学的背景下引入的,其中一个“操作分类学单位”只是目前正在研究的生物体群。在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。在生物信息分析中,一般来说,测序得到的每一条序列来自一个菌株。要了解一个样品测序结果中的菌种、菌属等数目信息,就需要对序列进行归类操作(cluster)。通过归类操作,将序列按照彼此的相似性分归为许多小组,一个小组就是一个OTU。
板块二:为何要进行OTU聚类?
测序完成后,每例样品的测序序列达到几万条,对每一条序列都可以进行物种注释,但该方式工作量大,每一条序列均需要与数据库进行比对且耗时,而且扩增、测序等过程中出现的错误会降低比对结果的准确性。
OTU聚类是将相似的微生物序列归类到同一个OTU中的过程。聚类可以根据序列相似性来组织微生物种类,从而更好地理解微生物群落的组成和结构。每个OTU代表了一组在某个相似性阈值下归类的微生物序列。通过聚类,可以将大量的微生物序列数据简化为一组代表性的OTU,从而减少计算和分析的复杂性。在16S多样性研究中,目前主要还是按照序列97%的相似性进行OTU聚类。主要原因:在16S全长比对中,97%相似性可以认定为同一个种,所以可以初步认为一个OTU属于一个种的微生物,而细菌16S研究中,解释度最可靠的分类学地位是“属”,所以97%相似度划分OTU可以被接受。如此操作,在简化了工作量的同时,还提高了分析效率,而且OTU在聚类过程中还可以去除一些错误的序列,如嵌合体序列,提高分析的准确性。
在测序数据下机后,我们要对数据进行统计,总的来说,单个样本数据量满足3万条tags就可以了,该数据量也是满足多样性分析要求,同时也满足文章发表要求的。这里需要注意的是测序平台的选择,有二代测序和三代测序之分。两者的区别在于三代是全长扩增子测序,包含了V1-V9区域,而二代测序则包含的是两个(如V4-V5区)或者单个(如V4区)区域。以二代测序(V4-V5)数据为例,以下展示的是样本二代测序序列长度主要集中在401bp至500bp。
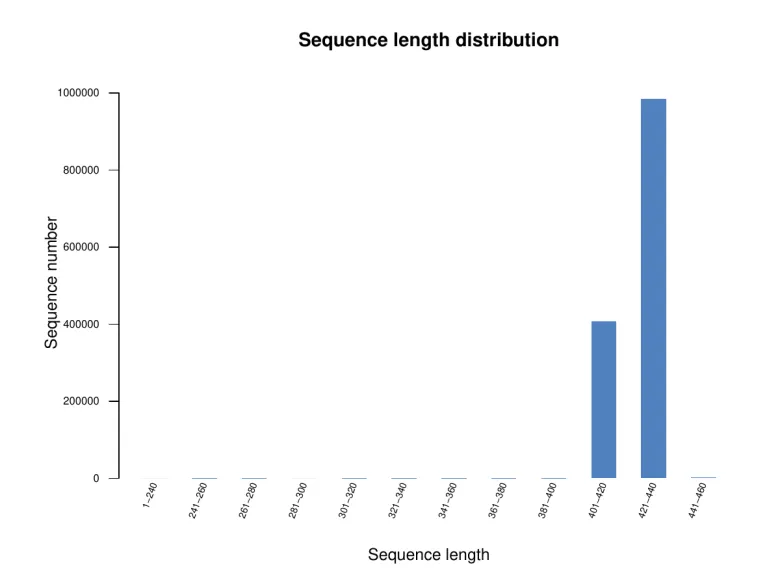
样本序列长度分布
在质控过滤完低质量数据后,对优化数据进行统计,然后就可以进行多样性的数据分析啦~
以下展示的是个样本序列统计表:

板块三:OTU聚类——分析方法
(1)聚类之前要对优化序列进行去冗余,便于降低分析中间过程冗余计算量;
(2)去除没有重复的单序列;
(3)按照97%相似性对非冗余序列(不含单序列)进行OTU聚类,在聚类过程中去除嵌合体,得到OTU的代表序列;
(4)将所有优化序列MAP至OTU代表序列,选出与OTU代表序列相似性在97%以上的序列,生成OTU表格,即OTU丰度表(OTU-table)。其中,OTU ID表示序列ID;第二列至后面的每一列,表示各样本序列在各个OTU中的丰度。

板块四:物种注释
为了得到每个OTU对应的物种分类信息,在OTU聚类完成后,会对每个OTU进行物种注释,采用RDPifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,生成OTU分类学综合信息表(OTU-taxa-table),并分别在各个分类水平:domain(域),kingdom(界),phylum(门),class(纲),order(目),family(科),genus(属),species(种)统计各样本的群落组成。其中,OTU ID表示序列ID;第二列至后面的每一列,表示各样本序列在各个OTU中的丰度,“taxonomy”表示最后一列可以查看分类学系谱信息,各级分类水平以“;”隔开,分类学名称前的单个字母为分类等级的首字母缩写,如domain(域)缩写为“d”,以“__”隔开。

在元莘生物的扩增子报告中,提供了OTU抽平和非抽平的结果文件。在抽平结果文件中包含有tax-summary-a和tax-summary-r两个文件夹,其中,-a文件夹中表示的是序列数的统计,-r文件夹中表示的是序列相对丰度百分比。这两个文件夹中的表格第一列均为物种分类名称,第一行均为样本名称,同时提供了不同分类水平的统计信息,分组或者不分组的。

