转自:生物谷
在我们的肠道里,住着一群小家伙——肠道细菌。有时候,它们中的某些“坏蛋”会过度生长,导致炎症性肠病或结直肠癌等疾病。然而,最近的一项研究发现,事情可能没有那么简单。来自欧洲分子生物学实验室(EuropeanMolecularBiologyLaboratory,EMBL)和哥本哈根大学的研究人员利用先进的机器学习算法,揭示了一个令人惊讶的事实:微生物载量(microbialload,即肠道中细菌的数量)的变化,而非疾病本身,可能是这些“坏蛋”细菌存在的主要原因。
相关研究结果于2024年11月14日在线发表在Cell期刊上,论文标题为“Fecalmicrobialloadisamajordeterminantofgutmicrobiomevariationandaconfounderfordiseaseassociations”。

论文的共同通讯作者、欧洲分子生物学实验室的PeerBork说:“我们非常惊讶地发现,许多以前被认为与疾病相关的微生物物种,实际上可以用微生物载量的变化来更好地解释。这表明这些微生物物种主要与腹泻和便秘等症状有关,而不是直接与疾病本身相关。”
克服技术难题
长期以来,微生物载量一直是微生物组研究中的一个重要因素,但由于实验方法的高成本和劳动密集型特点,大规模分析受到了限制。在这项研究中,研究人员利用机器学习方法克服了这一难题。他们根据微生物组的相对组成开发出一种粪便微生物载量预测模型,并将其应用于大规模宏基因组数据集,以探索这种载量在健康和疾病中的变化。
大规模数据的应用
论文的另一位共同通讯作者、欧洲分子生物学实验室的MichaelKuhn说:“测量粪便样本中的微生物载量需要大量努力,我们很高兴能访问两个大型宏基因组数据集,其中的微生物载量已经过实验测量。我们希望通过我们的方法,将这些数据推广到更大的领域,并利用我们提供的工具,预测所有成年人人类肠道微生物组研究中的微生物载量。”
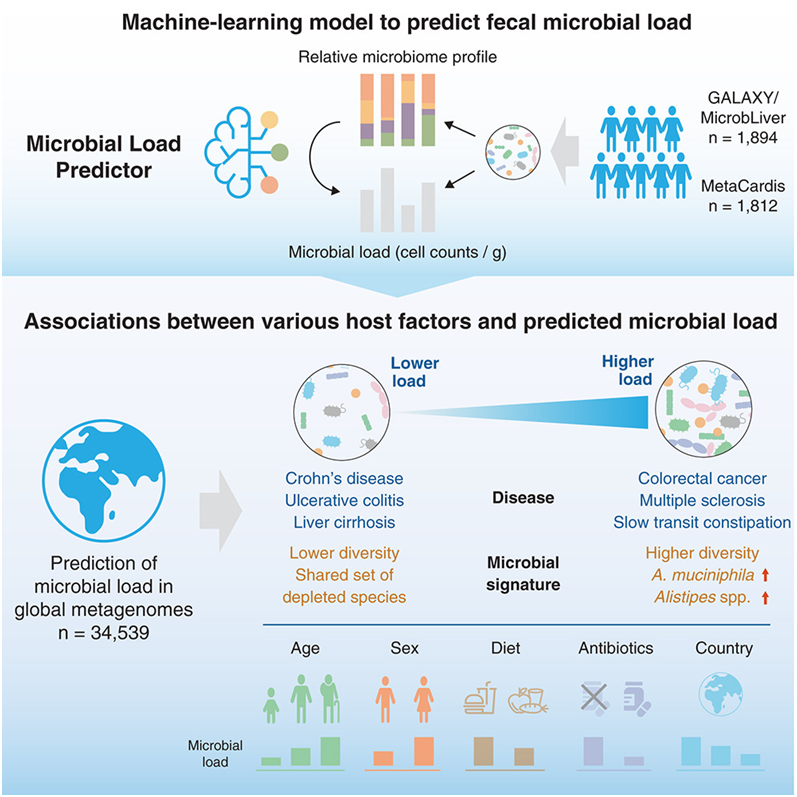
作者为这项研究生成的数据集包括成千上万个宏基因组,以及在GALAXY(Gut-and-LiverAxisinAlcoholicLiverFibrosis,酒精性肝纤维化中的肠-肝轴)和诺和诺德基金会的MicrobLiver项目中实验测量的微生物载量。他们还使用了之前公开的MetaCardis研究人群中的宏基因组和微生物载量数据。在探索性数据集方面,他们使用了以前研究中的数万个宏基因组,包括来自日本和爱沙尼亚的人群。
尽管这项研究提供了重要的见解,但作者也承认存在一些局限性。由于分析仅基于关联性,他们无法确定明确的因果关系,也无法提供机理方面的见解。此外,所开发的方法仅适用于人类肠道微生物组。要预测其他栖息地中的微生物载量,需要不同的训练数据集。
未来的研究将重点关注与疾病更直接相关的微生物物种,以便更好地了解它们在疾病病因学中的作用以及作为生物标志物的潜在用途。此外,将这一预测模型应用于其他环境,如海洋和土壤微生物组,可能进一步了解全球范围内的微生物生态学。
这项研究揭示了一个令人惊讶的事实:肠道中细菌的数量变化,而非疾病本身,可能是某些“坏蛋”细菌存在的主要原因。通过机器学习方法,研究人员克服了技术难题,为未来的研究提供了新的工具和思路。希望这些发现能帮助我们更好地理解肠道微生物组的复杂性,从而为疾病的预防和治疗提供新的途径。
参考资料:
SuguruNishijimaetal.Fecalmicrobialloadisamajordeterminantofgutmicrobiomevariationandaconfounderfordiseaseassociations.Cell,2024,doi:10.1016/j.cell.2024.10.022.
(转自:生物谷)
